Лекция № 12. Карбоновые кислоты
Лекция № 12
КАРБОНОВЫЕ КИСЛОТЫ
План
1. Методы получения.
2. Химические свойства.
2.1. Кислотные свойства.
2.2. Реакции нуклеофильного замещения.
Функциональные производные карбоновых кислот.
2.3. Реакции по a -углеродному атому.
2.4. Декарбоксилирование.
2.5. Восстановление.
2.6. Дикарбоновые кислоты.
Лекция № 12
КАРБОНОВЫЕ КИСЛОТЫ
План
1. Методы получения.
2. Химические свойства.
2.1. Кислотные свойства.
2.2. Реакции нуклеофильного замещения.
Функциональные производные карбоновых кислот.
2.3. Реакции по a -углеродному атому.
2.4. Декарбоксилирование.
2.5. Восстановление.
2.6. Дикарбоновые кислоты.
1. Методы получения
- Окисление первичных спиртов и альдегидов.
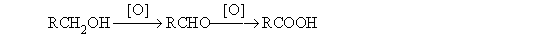
Окислители: KmnO4 в кислой или нейтральной среде, K2Cr2O7 в кислой среде.
- Окисление алканов.
RCH2CH2R’ + 5/2O2 ® RCOOH + R’COOH + H2O
Окисление осуществляют при катализе реакции солями
кобальта или марганца. - Окисление алкенов.

Окислители:
K2Cr2O7 или KMnO4 в кислой
среде. - Окисление алкилбензолов.
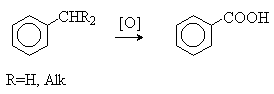
В качестве окислителей используют
KMnO4, Na2Cr2O7, кислород в присутствии солей Co и Mn. - Гидролиз нитрилов.
RCє N + 2H2O + HX ® RCOOH + NH4XRCє N + H2O + NaOH ® RCOONa + NH3
- Гидролиз производных карбоновых
кислот.
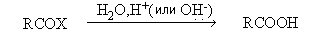
X= OR, Cl, OCOR, NH2
2. Химические
свойства
Карбоновые кислоты содержат карбоксильную группу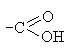 , в которой непосредственно связаны между
, в которой непосредственно связаны между
собой карбонильная группа и гидроксил. Их взаимное влияние обуславливает новый
комплекс свойств, отличных от свойств карбонильных соединений и
гидроксилпроизводных. Реакции с участием карбоновых кислот протекают по
следующим основным направлениям.
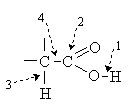
- Замещение водорода группы COОН под
действием оснований (кислотные свойства). - Взаимодействие с нуклеофильными реагентами
по карбонильному атому углерода (образование функциональных производных и
восстановление) - Реакции по a -углеродному атому
(галогенирование) - Декабоксилирование
2.1. Кислотные
свойства
Карбоновые кислоты – одни из самых сильных органических кислот. Их водные
растворы имеют кислую реакцию.
RCOOH + H2O = RCOO— +
H3O+
Причины высокой кислотности карбоновых кислот и
ее зависимость от природы заместителей в углеводородном радикале были
рассмотрены ранее (см. лек. №4).
Карбоновые кислоты образуют соли при
взаимодействии с активными металлами и большинством оснований.
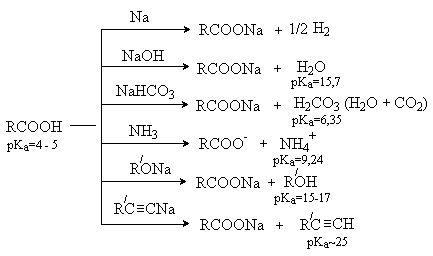
При взаимодействии с сильными неорганическими
кислотами карбоновые кислоты могут проявлять основные свойства, присоединяя
протон по карбонильному атому кислорода.
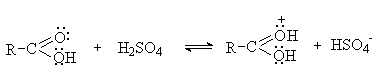
Протонирование карбоновых кислот используется
для активации карбоксильной группы в реакциях нуклеофильного замещения.
За счет присутствия в молекуле одновременно
кислотного и основного центров карбоновые кислоты образуют межмолекулярные
водородные связи и существуют в основном в виде димеров (см. лек. №2).
2.2. Реакции нуклеофильного замещения.
Функциональные производные карбоновых кислот.
Основной тип реакций карбоновых кислот –
взаимодействие с нуклеофилами с образованием функциональных производных.
Взаимопревращения, связывающие карбоновые кислоты и их функциональные
производные, приведены на схеме.
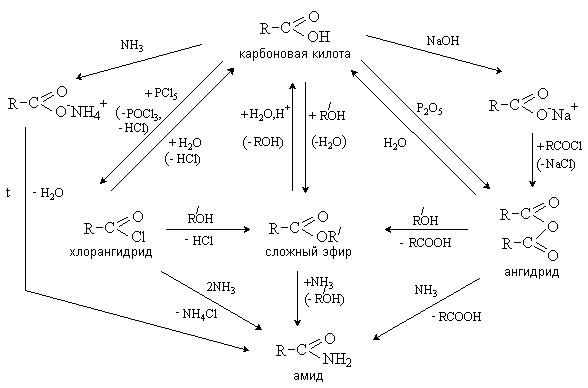
Представленные на схеме соединения содержат
ацильную группу ![]() В ходе
В ходе
их взаимопревращений она в неизменном виде переходит из одного соединения в
другое, соединяясь с нуклеофилом. Такие процессы называют ацилированием,
а карбоновые кислоты и их функциональные производные – ацилирующими
реагентами. В общем виде процесс ацилирования может быть представлен
следующей схемой.
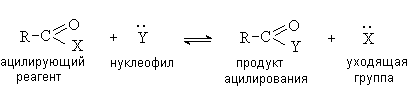
Таким образом, ацилирование представляет собой
процесс нуклеофильного замещения у карбонильного атома углерода.
Рассмотрим механизм реакции в общем виде и
сравним его с AdN-реакциями
альдегидов и кетонов. Как и в случае карбонильных соединений, реакция начинается
с атаки нуклеофила по карбонильному атому углерода, несущему эффективный
положительный заряд. При этом разрывается p -связь углерод-кислород и образуется тетраэдрический
интермедиат. Пути дальнейшего превращения интермедиата у карбонильных и
ацильных соединений различны. Если карбонильные соединения дают продукт присоединения, то ацильные соединения отщепляют группу X и дают продукт замещения.
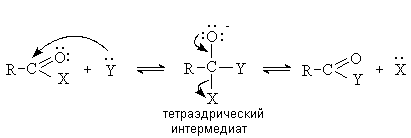
Причина разного поведения ацильных и
карбонильных соединений – в разной стабильности потенциальной уходящей группы X.
В случае альдегидов и кетонов это — гидрид-анион Н— или карбоанион R—, которые вследствие своей высокой основности являются
чрезвычайно плохими уходящими группами. В случае ацильных соединений Х
значительно более стабильная уходящая группа (Cl—,
RCOO—, RO—, NH2—), что делает возможным ее отщепление в виде аниона
Х— или сопряженной кислоты
НХ.
Реакционная способность по отношению к
нуклеофилам у карбоновых кислот и их функциональных производных меньше, чем у
альдегидов и кетонов, так как эффективный положительный заряд на карбонильном
атоме углерода у них ниже за счет + М- эффекта группы Х.
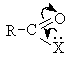
Активность ацильной группы повышается в условиях
кислотного катализа, так как при протонировании возрастает эффективный
положительный заряд на атоме углерода и облегчается его атака
нуклеофилом.
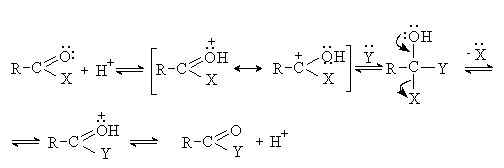
По ацилирующей способности производные
карбоновых кислот располагаются в следующий ряд в соответствии с уменьшением
+М-эффекта группы Х.
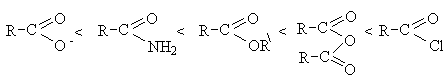
В этом ряду предыдующие члены могут быть получены из
последующих ацилированием соответствующего нуклеофила. Процесс получение более
активных ацилирующих реагентов из менее активных практически не идет из-за
неблагоприятного положения равновесия вследствие более высокой основности
уходящей группы по сравнению с атакующим нуклеофилом. Все функциональные
производные могут быть получены непосредственно из кислот и превращаются в них
при гидролизе.
Хлорангидриды и ангидриды
Методы получения
Хлорангидриды получают взаимодействием
карбоновых кислот с галогенидами фосфора и серы.
RCOOH + SOCl2 ® RCOOCl + SO2 +
HCl
RCOOH + PCl5 ® RCOOH + POCl3 +
HCl
Ангидриды образуются из карбоновых кислот под
действием оксида фосфора (V).
Cмешанные ангидриды могут быть получены
ацилированием солей карбоновых кислот хлорангидридами.
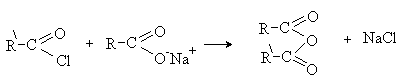
Реакции нуклеофильного замещения в
хлорангидридах и ангидридах.
Хлорангидриды и ангидриды — наиболее реакционноспособные производные
карбоновых кислот. Их реакции с нуклеофилами протекают в мягких условиях, без
катализатора и практически необратимо.
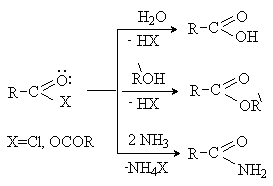
При использовании смешанных ангидридов с
нуклеофилом соединяется остаток более слабой кислоты, а анион более сильной
кислоты играет роль уходящей группы.
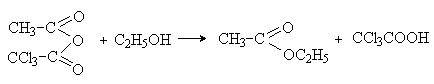
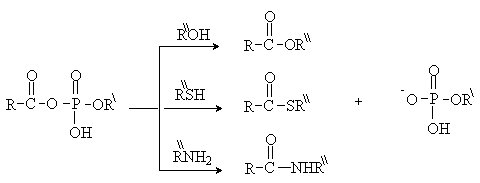
В
биохимических реакциях ацилирования важную роль играют смешанные ангидриды
карбоновых кислот и фосфорной кислоты – ацилфосфаты и замещенные ацилфосфаты. С
нуклеофилом соединяется остаток органической кислот, а ацилфосфат-анион
выполняет роль хорошей уходящей группы.
Сложные эфиры
Методы получения
- Из карбоновых кислот и спиртов (реакция этерификации).
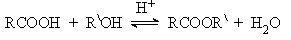
- Из хлорангидридов или ангидридов и
спиртов.
RCOX + ROH ® RCOOR + HX
X=Cl, OCOR - Переэтерификация.

- Из солей карбоновых кислот и
алкилгалогенидов.
карбоксильной группе.
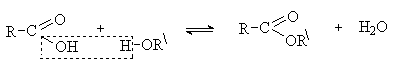
Карбоновые кислоты являются слабыми ацилирующими
реагентами из-за значительного +М-эффекта группы ОН. Использование сильных
нуклеофилов, которые одновременно являются и сильными основаниями (например,
основной катализ), в данном случае невозможно, так как они переводят карбоновые
кислоты в еще менее реакционноспособные соли карбоновых кислот. Реакцию проводят
в условиях кислотного катализа. Роль кислотного катализатора состоит, как уже
говорилось, в увеличении эффективного положительного заряда на атоме углерода
карбоксильной группы, и, кроме того, протонирование ОН группы на стадии
отщепления превращает ее в хорошую уходящую группу – Н2О.
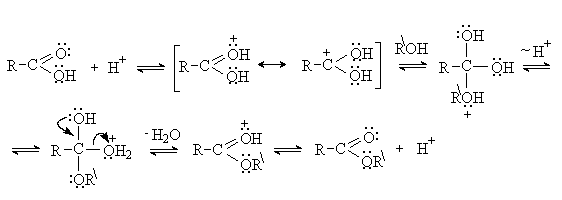
Все стадии реакции этерификации
обратимы. Для смешения равновесия в сторону процесса этерификации используют
избыток одного из реагентов или удаление продуктов из сферы реакции.
Реакции нуклеофильного замещения в
алкоксикарбонильной группе.
Сложные эфиры – более слабые ацилирующие
реагенты, чем ангидриды и хлорангидриды. SN-реакции в алкоксикарбонильной группе протекают в более
жестких условиях и требуют кислотного или основного катализа. Важнейшими
реакциями этого типа являются гидролиз, аминолиз и
переэтерификация.
Гидролиз.
Сложные эфиры гидролизуются с образованием карбоновых кислот под действием
кислот или щелочей.
Кислотный гидролиз сложных эфиров – это реакция обратная этерификации.
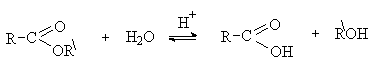
Механизм кислотного гидролиза включает те же стадии, что
и процесс этерификации, но в обратной последовательности.
Щелочной гидролиз сложных эфиров требует
эквимолярных количеств щелочи и протекает необратимо.
RCOOR + NaOH ® RCOO—Na+ + ROH
Суть щелочного катализа состоит в использовании
вместо слабого нуклеофила — воды, более сильного нуклеофила –
гидроксид-иона.
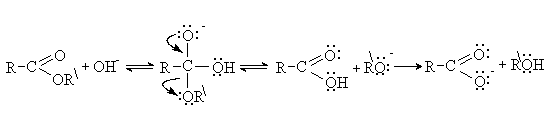
Необратимость процесса
обеспечивается низкой реакционной способностью по отношению к нуклеофилам
продукта гидролиза – карбоксилат-аниона.
Переэтерификация.
В реакции переэтерификации роль нуклеофила
выполняет молекула спирта. Процесс катализируется кислотами или
основаниями.
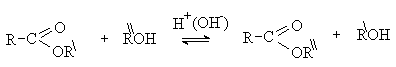
Механизм реакции аналогичен гидролизу сложных
эфиров. Переэтерификация – обратимый процесс. Для сдвига равновесия вправо
необходимо использовать большой избыток исходного спирта. Реакция
переэтерификации находит применение для получения сложных эфиров жирных кислот
из триацилглицеридов (см. лек. 18)
Аминолиз.
Сложные эфиры ацилируют аммиак и амины с
образованием амидов карбоновых кислот.
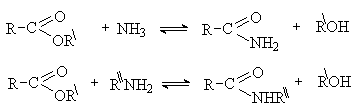
Амиды карбоновых кислот
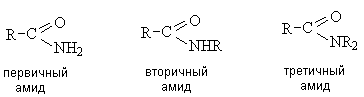
Строение амидной группы
Амидная группа встречается во многих биологически важных соединениях,
прежде всего в пептидах и белках (пептидная связь). Её электронное и
пространственное строение во многом определяет их биологическое
функционирование.
Амидная группа представляет собой р-p -сопряженную систему, в которой происходит
дополнительное перекрывание р-орбитали атома азота с p -орбиталью связи
углерод-кислород.
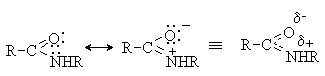
Такое распределение электронной плотности
приводит к увеличению энергетического барьера вращения вокруг связи С-N до 60 –
90 кДж/моль. В результате амидная связь имеет плоское строение, а длины связей
C-N и С=О имеют значения соответственно меньше и больше своих обычных
величин.
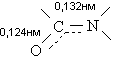
Отсутствие свободного вращения вокруг связи C-N
приводит к существованию у амидов цис- и транс-изомеров. Для
большинства амидов предпочтительной является транс-конфигурация.
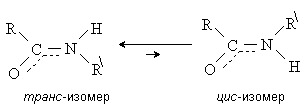
Пептидная связь также имеет транс-конфигурацию, в которой боковые радикалы аминокислотных остатков
наиболее удалены друг от друга
Методы получения
- Ацилирование аммиака или аминов
хлорангидридами или ангидридами.RCOX + 2NH3 ® RCONH2 +
NH4XRCOX + 2RNH2 ® RCONHR + RNH3X
X=Cl, OCOR
- Аминолиз сложных эфиров.
RCOOR + NH3 ® RCONH2 + ROHRCOOR + R\NH2 ® RCONHR\ + ROH
- Из карбоновых кислот и аммиака или
аминов.
RCOOH + NH3 ® RCOO—NH4+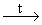 RCOONH2 +
RCOONH2 +
H2ORCOOH + RNH2 ® RCOONH3R RCOONHR + H2O
- Гидролиз нитрилов.
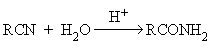
Реакции нуклеофильного замещения в
карбоксамидной группе.
Амиды — наименее реакционноспособные производные карбоновых кислот. Для них
известны реакции гидролиза, которые протекают в жестких условиях под действием
водных растворов кислот или щелочей.
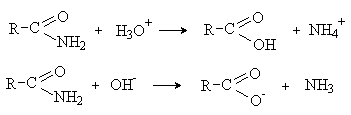
Механизмы реакций аналогичны гидролизу сложных
эфиров. Однако, в отличие от гидролиза эфиров, кислотный и щелочной гидролиз
амидов протекают необратимо.
2.3. Реакции по a -углеродному
атому
Карбоновые кислоты, содержащие a -водородные атомы,
взаимодействуют с бромом в присутствии фосфора с образованием исключительно a -бромпроизводных
(реакция Гелля – Форгальда — Зелинского)
![]()
Галоген в a -галогензамещенных кислотах легко замещается под
действием нуклеофильных реагентов. Поэтому a -галогензамещенные кислоты
являются исходными веществами в синтезе широкого круга замещенных по a -положению
кислот, в том числе a -амино- и a -гидроксикислот.
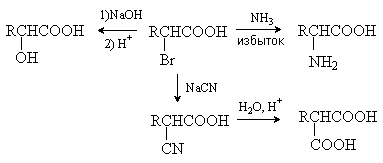
2.4.
Декарбоксилирование
Декарбоксилирование – это элиминирование CO2 из карбоновых кислот или их солей. Декарбоксилирование
проводят путем нагревания в присутствии кислот или оснований. При этом, как
правило, происходит замещение карбоксильной группы на атом водорода.
Незамещенные монокарбоновые кислоты
декарбоксилируются в жестких условиях.
![]()
Декарбоксилирование облегчается при наличии
электроноакцепторных заместителей в a -положении.
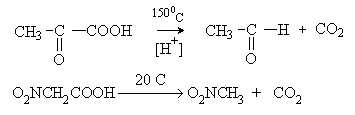
Важное значение имеет ферментативное
декарбоксилирование кето-, амино- и гидроксикислот в организме ( см. лек. №14 и
16).
Декарбоксилирование путем нагревания (сухой
перегонки) кальциевых и бариевых солей карбоновых кислот – метод получения
кетонов.
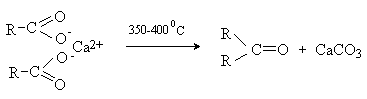
2.5.
Восстановление.
Карбоновые кислоты, хлорангидриды, ангидриды и сложные эфиры
восстанавливаются LiAlH4 до первичных
спиртов.

Хлорангидриды могут быть восстановлены до
альдегидов (см. лек. № 11).
При восстановлении амидов карбоновых кислот
образуются амины.
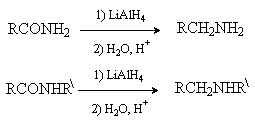
3. Дикарбоновые кислоты
Дикарбоновые кислоты содержат две карбоксильные группы. Наиболее доступными
являются кислоты линейного строения, содержащие от 2 до 6 атомов углерода. Их
строение и методы получения представлены в таблице 9.
Таблица 9. Методы получения и свойства дикарбоновых кислот.
Формула |
Название |
рКа1 |
Метод получения |
HOOC-COOH |
Щавелевая (этандиовая) |
1,23 |
|
HOOCCH2COOH |
Малоновая (1,3-пропандиовая) |
2,80 |
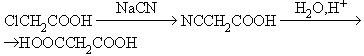 |
HOOC(CH2)2COOH |
Янтарная (1,4-бутандиовая) |
4,17 |
|
HOOC(CH2)3COOH |
Глутаровая (1,5-пентандиовая) |
4,34 |
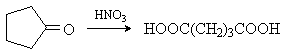 |
HOOC(CH2)4COOH |
Адипиновая (1,6-гександиовая) |
4,43 |
|
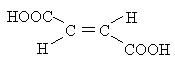 |
Фумаровая (транс-1,4-бутендиовая) |
3,03 |
Из глюкозы под действием бактерий |
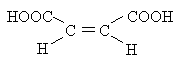 |
Малеиновая (цис-1,4-бутендиовая) |
1,93 |
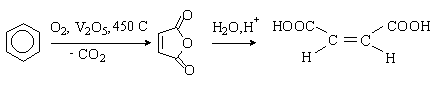 |
Химические свойства дикарбоновых кислот в
основном аналогичны свойствам монокарбоновых кислот. Они дают все реакции,
характерные для карбоксильной группы. При этом могут быть получены
функциональные производные (хлорангидриды, ангидриды, сложные, эфиры, амиды) как
по одной, так и по обеим карбоксильным
группам. Дикарбоновые кислоты имеют большую кислотность, чем монокарбоновые,
вследствие –I-эффекта карбоксильной группы. По мере увеличения расстояния между
карбоксильными группами кислотность дикарбоновых кислот уменьшается (см. табл.
9).
Кроме того, дикарбоновые кислоты имеют ряд
специфических свойств, которые определяются наличием в молекуле двух
карбоксильных групп.
Отношение дикарбоновых кислот к
нагреванию.
Превращения дикарбоновых кислот при нагревании
зависят от длины цепи, разделяющей карбоксильные группы, и определяются
возможностью образования термодинамически стабильных пяти- и шестичленных
циклов.
При нагревании щавелевой и малоновой кислот
происходит декарбоксилирование.
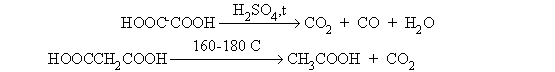
Янтарная, глутаровая и малеиновая кислоты при
нагревании легко отщепляют воду с образованием пяти- и шестичленных циклических
ангидридов.
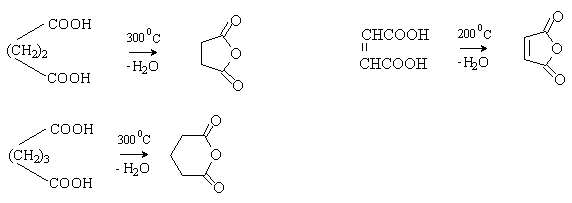
Адипиновая кислота при нагревании
декарбоксилируется с образованием циклического кетона – циклопентанона.
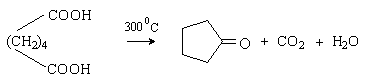
Реакции поликонденсации
Дикарбоновые кислоты взаимодействуют с диаминами и диолами с
образованием соответственно полиамидов и полиэфиров, которые используются в
производстве синтетических волокон.
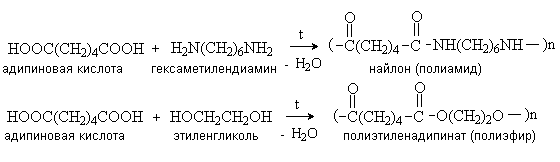
Биологически важные дикарбоновые
кислоты.
Щавелевая кислота образует труднорастворимые соли, например,
оксалат кальция, которые отлагаются в виде камней в почках и мочевом пузыре.
Янтарная кислота участвует в обменных процессах, протекающих в
организме. Является промежуточным соединением в цикле трикарбоновых кислот.
Фумаровая кислота, в отличие от малеиновой, широко распространена в природе, участвует в процессе
обмена веществ, в частности в цикле трикарбоновых кислот.
NO
Не скачивается.
скачивается.